

La maladie de Charcot-Marie-Tooth (CMT) est souvent confondue avec la maladie de Charcot (SLA), alors que ce sont des maladies très différentes.

Les maladies de Charcot-Marie-Tooth (CMT en abrégé) sont les maladies neuromusculaires héréditaires les plus fréquentes. Elles doivent leur nom aux trois médecins qui les ont décrites en 1886 : Jean-Martin Charcot, Pierre Marie, et Howard Henry Tooth.
Elles sont aussi appelées neuropathies périphériques héréditaires sensitives et motrices, pour les distinguer de la maladie de Charcot ou Sclérose Latérale Amyotrophique (SLA).
1 personne sur 2 500 soit environ 40 000 personnes en France en sont atteintes, sans distinction de sexe ni d’âge (début dans l’enfance ou chez l’adulte).
Les CMT sont des maladies génétiques, héréditaires, neuromusculaires, évolutives, qui n’entament pas l’espérance de vie. Elles atteignent les nerfs périphériques provoquant essentiellement une amyotrophie des mollets, des avant-bras et des mains.
L’âge d’apparition des symptômes est très variable, le plus souvent dans l’enfance ou chez l’adulte jeune.
Les premiers signes concernent généralement les pieds qui se creusent, deviennent insensibles et peu stables. Peu à peu une gêne à la marche (steppage) s’installe provoquant des chutes, des entorses, une difficulté à courir et une diminution du périmètre de marche. Des rétractions tendineuses peuvent provoquer la mise en « griffe » des orteils. Une fonte musculaire (amyotrophie) peut apparaître et donner un aspect caractéristique en “mollet de coq ».
L’atteinte des mains n’est pas systématique et apparaît généralement après plusieurs années d’évolution : les mouvements fins sont difficiles à exécuter, les doigts peuvent se mettre en « griffe », l’amyotrophie peut provoquer la perte de la fonction « de pince » (pouce index) et une diminution de force. En principe toutes ces atteintes sont bilatérales.
Les déformations et l’amyotrophie sont variables. En revanche certains symptômes sont fréquents : fatigue, équilibre instable, station debout pénible, montée d’escaliers difficile et habileté manuelle diminuée. Les crampes sont fréquentes surtout en période d’évolution.
Bien que la CMT n’affecte pas l’espérance de vie, le niveau de handicap est très variable et peut aller d’une légère difficulté à la marche jusqu’à la nécessité d’une canne ou d’un fauteuil roulant.
Les CMT progressent généralement de manière lente, toutefois, elles peuvent connaître des phases d’accélération, notamment durant l’adolescence, en période de grossesse, à la ménopause, ou dans des situations stressantes.
L’évolution de la maladie dépend du type de CMT, mais aussi de chaque individu, rendant difficile la prévision de son évolution. La sévérité des symptômes varie d’une personne à l’autre, y compris au sein d’une même famille.


Les maladies de Charcot-Marie-Tooth sont génétiques, héréditaires (il existe aussi des cas sporadiques, dits de novo). Les anomalies génétiques en cause concernent des gènes impliqués dans la production de protéines qui entrent dans la constitution ou le fonctionnement des nerfs.
À l’heure actuelle, plus de 130 gènes ont été identifiés comme pouvant être responsables d’une maladie de Charcot-Marie-Tooth.
Il existe différentes formes de CMT, selon la nature de l’atteinte du nerf périphérique (forme axonale, forme démyélinisante et forme intermédiaire). Le chiffre désigne le type d’atteinte et son mode de transmission, la lettre détermine le gène.

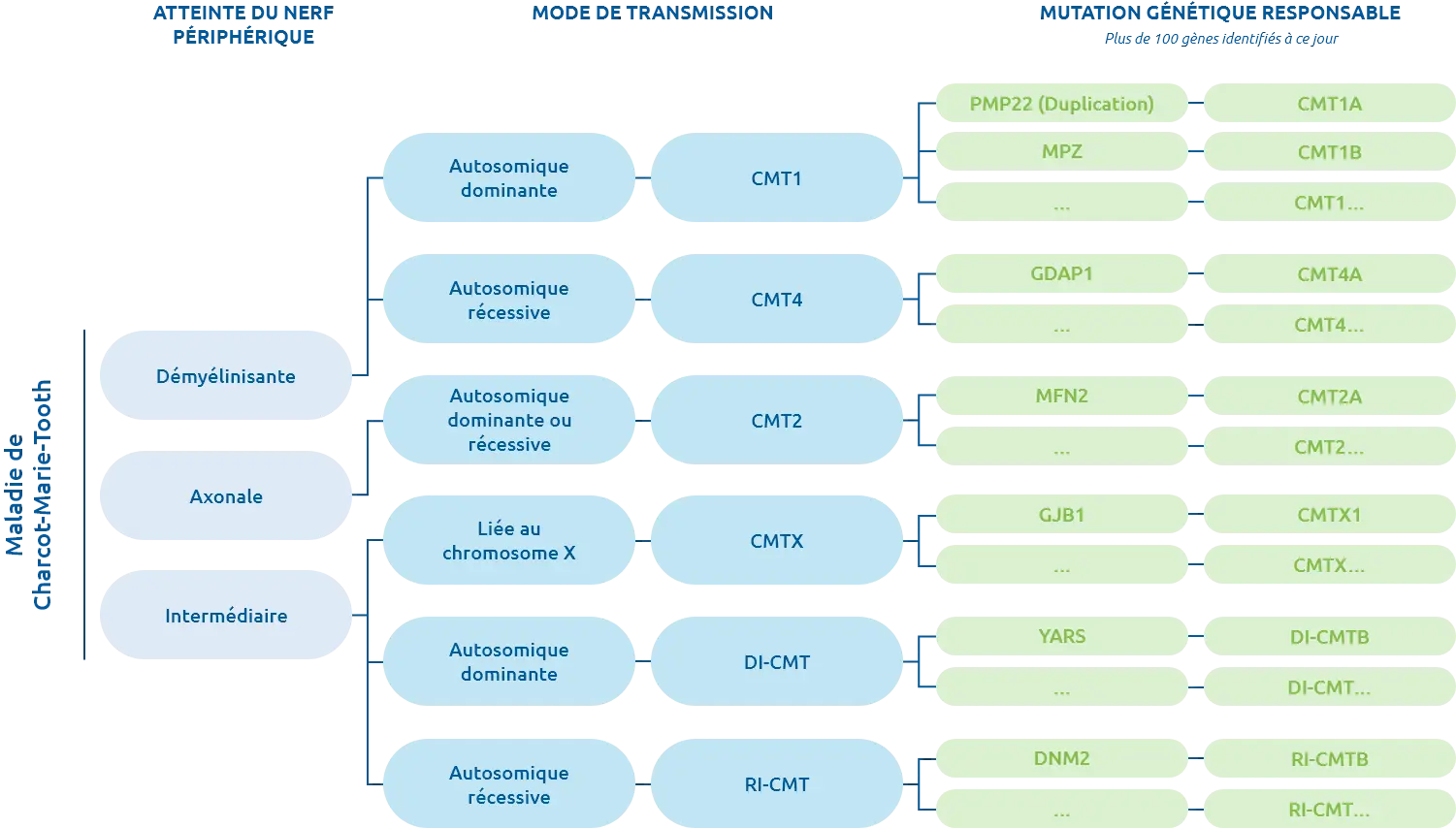
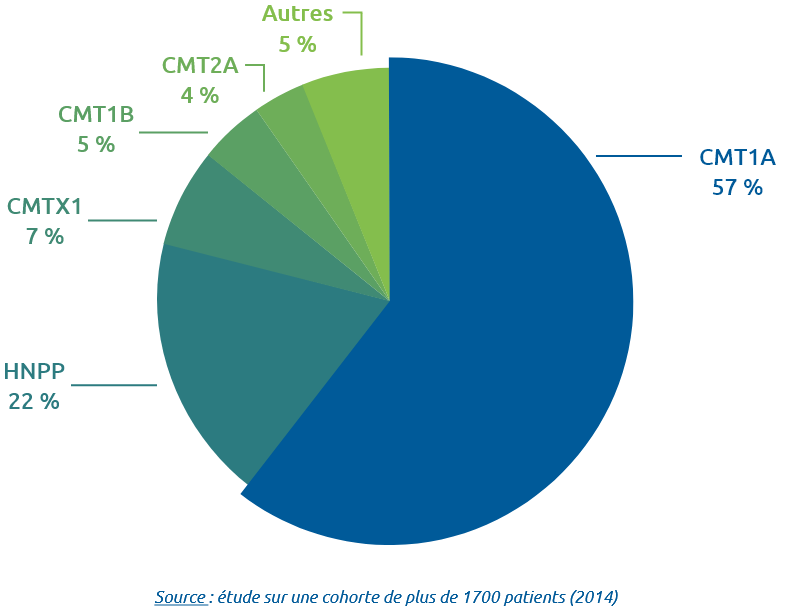
Certaines formes, comme la CMT1A la plus fréquente, présentent une altération de la gaine de myéline qui protège et entoure les axones du nerf (formes démyélinisantes). D’autres formes comme les CMT2, affectent directement l’axone du nerf (formes axonales). Certaines, comme la CMTX, sont des formes intermédiaires (à la fois démyélinisantes et axonales). L’appellation CMT3 a été utilisée, puis abandonnée.
La gaine de myéline est produite par les cellules de Schwann, cellules indispensables à la survie et à la maturation des cellules nerveuses, les neurones.
Nerf en bonne santé vs nerf cmt


Une autre maladie proche de la CMT est appelée Tomacula ou Neuropathie Héréditaire avec Hypersensibilité à la Pression. Elle fait également partie des neuropathies sensitivo-motrices héréditaires.
La neuropathie tomaculaire est une neuropathie périphérique héréditaire associée aux CMT, même si la nomenclature scientifique et médicale en France ne la considère pas pour le moment comme une CMT à proprement parler. Malgré tout elle est « cousine » des CMT1A, car c’est la même protéine de la myéline (PMP22) qui est impliquée dans ces deux maladies. La neuropathie héréditaire est le plus souvent associée à une disparition (délétion) des informations génétiques codant pour PMP22 sur un des deux chromosomes alors que ces informations sont, au contraire, dupliquées dans la maladie de Charcot-Marie-Tooth de type CMT1A. Ainsi les malades atteints de neuropathie tomaculaire produisent moins de PMP22 que la normale alors que ceux atteints de CMT1A en produisent plus.
Après compression d’un nerf, ces malades développent une neuropathie qui peut durer plusieurs mois. La récupération est, ensuite, pour la majorité des cas, très souvent complète.
Elle se manifeste le plus souvent par une perte de sensibilité et une faiblesse musculaire qui apparaissent soudainement, généralement après un stress du nerf comme une compression, un étirement ou un mouvement répétitif. D’autres signes peuvent se présenter : pieds tombants, faiblesse dans les bras et les mains, engourdissements… Fatigue et douleur sont fréquemment rapportées. Dans la grande majorité des cas, les patients récupèrent en quelques semaines ou mois, mais, dans certains cas, les troubles sensoriels et moteurs ne disparaissent pas.
Les troubles répétés sur le même nerf peuvent provoquer des lésions durables. La prévention des compressions du nerf est donc nécessaire pour éviter les épisodes.
Le nombre important de gènes impliqués dans les maladies de Charcot-Marie-Tooth et l’hétérogénéité clinique rendent la recherche de pistes thérapeutiques complexe.
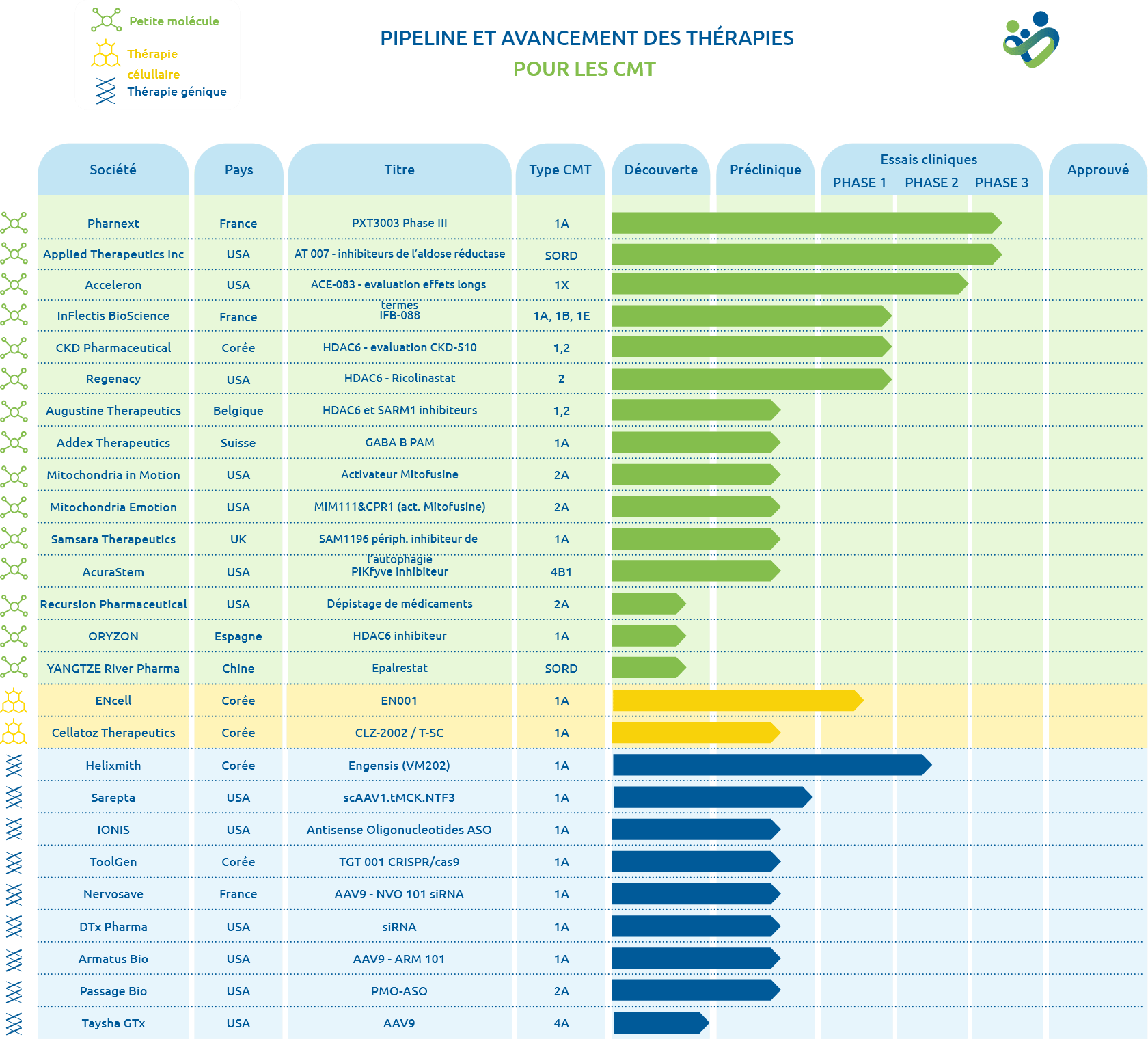
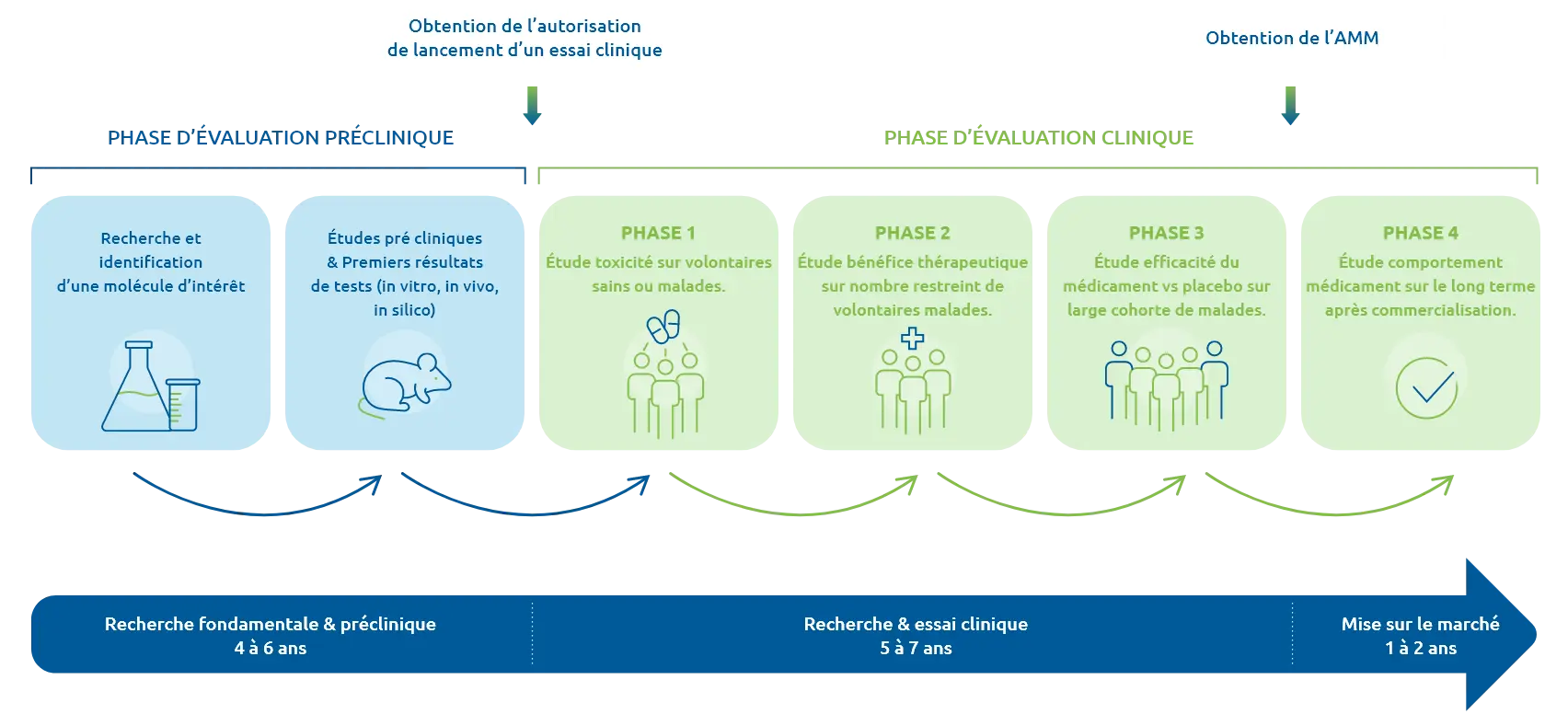
Découvrez les phases de développement
d'un médicament
Une prise en charge globale :
du diagnostic au suivi
Les centres de référence maladies rares (CRMR) rassemblent une équipe hospitalière spécialisée experte sur une maladie rare ou un groupe de maladies rares.
+ Cette équipe est médicale, mais intègre aussi des compétences paramédicales, psychologiques, médico-sociales, éducatives, sociales, et des partenariats avec les associations de personnes malades.
Les centres de compétence (CCMR) assurent la prise en charge et le suivi des patients au plus proche de leur domicile, en lien avec le réseau de CRMR dont ils dépendent.
+ Ils rassemblent une équipe hospitalière spécialisée experte sur une maladie rare ou un groupe de maladies rares. Ils font le lien avec les professionnels de santé hospitaliers ou de ville, et les secteurs du social, éducatif, médico-social sur leurs territoires de santé. Ils facilitent aussi l’accès aux dispositifs, droits et prestations dédiés aux personnes handicapées et à leurs aidants, en partenariat avec les MDPH notamment.
Pour trouver le centre de référence ou de compétence le plus proche de chez vous, veuillez contacter votre délégué de région.
Au fil des années, CMT-France a contribué à la réalisation d’un ensemble de supports thématiques autour de la CMT.
Ressources essentielles pour mieux comprendre la maladie, en améliorer la prise en charge, partager et mieux vivre avec, veuillez contacter votre délégué de région pour vous en procurer.
Vous trouverez ci-dessous des extraits de brochures, livrets, BD…
Découvrez notre boutique en ligne pour vous procurer la BD « Timéo, un collégien comme les autres » : cliquez ici
Découvrez notre boutique en ligne pour vous procurer le livre « Rencontres avec des personnes rares » : cliquez ici
Le PNDS pour les CMT est un document de référence de bonnes pratiques portant spécifiquement sur les maladies de Charcot-Marie-Tooth.
+ L’objectif du PNDS est d’expliciter aux médecins spécialistes la prise en charge diagnostique et thérapeutique optimale, ainsi que le parcours de soins d’un patient CMT. Une synthèse existe pour les médecins traitants.
La prise en charge précoce en médecine physique et réadaptation des patients CMT est indispensable pour limiter les conséquences de la maladie et tenter de ralentir son évolution.
+ Elle comprend la kinésithérapie pour notamment préserver la qualité de l’équilibre, prévenir les chutes, limiter la fatigue à la marche, maintenir un périmètre de déplacement satisfaisant, conserver une endurance correcte. La prise en charge rééducative est propre à chaque malade.
L’auto-rééducation réalisée quotidiennement ou très régulièrement est un facteur déterminant pour prolonger les effets de la rééducation et maintenir sa qualité de vie.
Le sport a été longtemps décrié, or la pratique régulière d’une Activité Physique Adaptée (APA) pour les personnes atteintes de la CMT est fortement conseillée et présente de nombreux bénéfices.
+ Dans une approche personnalisée à chaque patient, le sport et l’APA améliorent la qualité de vie, particulièrement en luttant contre la sédentarité et en renforçant les capacités physiques. Il existe une très grande variété de pratiques sportives et d’Activités Physiques Adaptées, ainsi chacun peut trouver celle qui lui convient le mieux et lui apporte le plus de plaisir. Cette notion de plaisir est cruciale pour une pratique sur le long terme.
Il existe de nombreux professionnels et structures qui proposent de l’Activité Physique Adaptée.
L’appareillage (orthèses, releveurs…) représente l’aspect palliatif de la prise en charge.
+ La prescription d’appareillage du membre inférieur se fait après l’analyse des troubles patho-mécaniques du déplacement (la marche et montée/descente des escaliers) et de la station debout et leur aggravation par la douleur. Selon les cas, la chirurgie fonctionnelle des déformations neuro-orthopédiques du pied permet aussi d’obtenir des résultats satisfaisants.
L’ergothérapeute accompagne les personnes en situation de handicap et propose des programmes de rééducation, modifie la façon de réaliser une activité avec ou sans aides techniques et participe à aménager l’environnement.
+ Son rôle vise à améliorer, à maintenir ou à suppléer les capacités motrices, cognitives, psychologiques et/ou sociales. Il contribue à une meilleure autonomie dans les activités de la vie quotidienne.
Les cures thermales « neurologie » propose une prise en charge thérapeutique globale pour les patients porteurs de certaines affections neuromusculaires.
+ Les symptômes associés, comme les douleurs musculo-squelettiques, les troubles de la motricité, la fatigue… peuvent être améliorés par les cures thermales. Elles soulagent les douleurs, favorisent la mobilité, freinent la consommation de médicaments et améliorent le quotidien des malades.
Une alimentation équilibrée contribue à une bonne santé, un état de bien-être et favorise une croissance harmonieuse chez l’enfant. Chez les malades CMT, l’équilibre alimentaire, dont le contrôle du surpoids, aide à mieux compenser les conséquences de la maladie (douleurs et fatigue) et à se sentir mieux physiquement et moralement.
La douleur chronique est une composante importante dans la CMT, mais difficile à prendre en charge. Elle requiert souvent de consulter des spécialistes de la douleur dans une approche pluridisciplinaire.
La fatigue dans les CMT peut s’expliquer par plusieurs mécanismes dont la faiblesse musculaire et la douleur. Elle affecte le quotidien du malade et influence sa capacité à produire une performance, qu’elle soit physique ou cognitive.
Focus / avertissement
La balance bénéfices/risques
Avant de proposer un traitement, le médecin évalue la balance bénéfices/risques pour son patient. Ce dernier doit lui aussi l’évaluer pour lui-même avant de prendre une décision.
Comme son nom l’indique, la balance bénéfices/risques consiste à mettre en balance d’un côté les bénéfices escomptés et de l’autre les risques connus et/ou attendus d’un traitement, d’un soin ou d’un examen. Pour que ce dernier soit envisagé, il est nécessaire que la balance penche en faveur du ou des bénéfices.
La balance bénéfices/risques repose sur un ensemble d’éléments qui doivent être pris en compte tant par le médecin que par le patient, dans le cadre d’un dialogue nécessaire à une décision véritablement partagée.
De plus, cette balance est susceptible d’évoluer, en fonction de l’évolution des connaissances scientifiques, mais aussi de l’évolution de la situation du patient. Elle doit donc être régulièrement réévaluée.
Découvrez notre boutique en ligne pour vous procurer la BD « Timéo, un collégien comme les autres : cliquez ici
Adhérez pour unir nos forces et agir ensemble
contre la maladie de Charcot-Marie-Tooth !

